Nanoxis Consulting AB
Microfluidic flow cell for sequential digestion of immobilized proteoliposomes
Jansson ET, Trkulja CL, Olofsson J, Millingen M, Wikström J, Jesorka A, Karlsson A, Karlsson R, Davidson M, Orwar O.Anal Chem. 2012 Jul 3;84(13):5582-8. doi: 10.1021/ac300519q. Epub 2012 Jun 13.
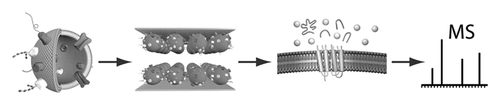
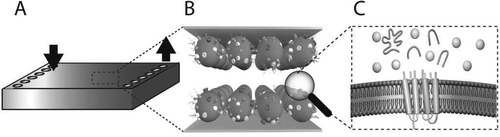
Abstract
We have developed a microfluidic flow cell where stepwise enzymatic digestion is performed on immobilized proteoliposomes and the resulting cleaved peptides are analyzed with liquid chromatography-tandem mass spectrometry (LC-MS/MS). The flow cell channels consist of two parallel gold surfaces mounted face to face with a thin spacer and feature an inlet and an outlet port. Proteoliposomes (50-150 nm in diameter) obtained from red blood cells (RBC), or Chinese hamster ovary (CHO) cells, were immobilized on the inside of the flow cell channel, thus forming a stationary phase of proteoliposomes. The rate of proteoliposome immobilization was determined using a quartz crystal microbalance with dissipation monitoring (QCM-D) which showed that 95% of the proteoliposomes bind within 5 min. The flow cell was found to bind a maximum of 1 μg proteoliposomes/cm(2), and a minimum proteoliposome concentration required for saturation of the flow cell was determined to be 500 μg/mL. Atomic force microscopy (AFM) studies showed an even distribution of immobilized proteoliposomes on the surface. The liquid encapsulated between the surfaces has a large surface-to-volume ratio, providing rapid material transfer rates between the liquid phase and the stationary phase. We characterized the hydrodynamic properties of the flow cell, and the force acting on the proteoliposomes during flow cell operation was estimated to be in the range of 0.1-1 pN, too small to cause any proteoliposome deformation or rupture. A sequential proteolytic protocol, repeatedly exposing proteoliposomes to a digestive enzyme, trypsin, was developed and compared with a single-digest protocol. The sequential protocol was found to detect ~65% more unique membrane-associated protein (p < 0.001, n = 6) based on peptide analysis with LC-MS/MS, compared to a single-digest protocol. Thus, the flow cell described herein is a suitable tool for shotgun proteomics on proteoliposomes, enabling more detailed characterization of complex protein samples.
Strain-level typing and identification of bacteria using mass spectrometry-based proteomics
Karlsson R, Davidson M, Svensson-Stadler L, Karlsson A, Olesen K, Carlsohn E, Moore ER.J Proteome Res. 2012 May 4;11(5):2710-20. doi: 10.1021/pr2010633. Epub 2012 Apr 11.
Abstract
Because of the alarming expansion in the diversity and occurrence of bacteria displaying virulence and resistance to antimicrobial agents, it is increasingly important to be able to detect these microorganisms and to differentiate and identify closely related species, as well as different strains of a given species. In this study, a mass spectrometry proteomics approach is applied, exploiting lipid-based protein immobilization (LPI), wherein intact bacterial cells are bound, via membrane-gold interactions, within a FlowCell. The bound cells are subjected to enzymatic digestion for the generation of peptides, which are subsequently identified, using LC-MS. Following database matching, strain-specific peptides are used for subspecies-level discrimination. The method is shown to enable a reliable typing and identification of closely related strains of the same bacterial species, herein illustrated for Helicobacter pylori .
Membrane protein profiling of human islets of Langerhans using several extraction methods
Hansson SF, Henriksson Å, Johansson L, Korsgren O, Eriksson JW, Tornqvist H, Davidsson P.
Clin. Proteom., 2010, 6(4), 195 - 207.
Introduction
Proteomic characterization of the human pancreatic islets, containing the insulin producing beta-cells, is likely to be of great importance for improved treatment and understanding of the pathophysiology of diabetes mellitus.
Objective
The focus of this study was to characterize the human islet membrane proteome.
Methods
In order to identify as many membrane proteins as possible, five different extraction procedures were used, i.e., phase separation using Triton X-114, a plasma membrane protein kit, cell surface protein biotinylation, total protein extraction, and lipid-based protein immobilization flow cell. Digested protein extracts were analyzed by nanoflow liquid chromatography tandem mass spectrometry. Then the identified proteins were categorized according to cellular location using their gene ontology annotation and by prediction of transmembrane helices in the sequence. This information was used to estimate the amount of membrane proteins identified.
Results
By combining the results from all extraction procedures, the total number of membrane proteins identified from the human islets was increased, accentuating that a combination of methods usually gives a higher coverage of the proteome. A total of 1,700 proteins were identified (≥2 unique peptides), and 735 of these proteins were annotated as membrane proteins while 360 proteins had at least one predicted transmembrane helix. The extraction method using phase separation with Triton X-114 yielded both the highest number and the highest proportion of membrane proteins.
Conclusion
This study gave an enhanced characterization of the human islet membrane proteome which may contribute to a better understanding of islet biology.